 Open Access
Open Access Abstract
This perspective reviews the effects of different tissue engineering techniques, for induced differentiation of human embryonic stem cells into pulmonary lineage specific cells, and validating cell based regenerative therapy to ameliorate pulmonary degeneration. The etiology of degeneration differs from disease to disease. Likewise, strategies to regenerate lost tissues in its heterogeneity, should also cater to the regeneration process. To customize the same, researchers ought to device strategies to repair, replace or regenerate, in the spatio- temporal format, healthy tissues in their heterogeneous multi-functional existence, such that when transplanted in vitro, they may find their niche and integrate seamlessly into the tissue system and manifest full-fledged functional competence in order to reverse the erstwhile effects of degeneration. Cell based lung repair or regeneration is unquestionably the most promising agenda of regenerative medicine.
In one of our studies, human embryonic stem cell (hESC) line H7 was successfully differentiated into three non-ciliated alveolar epithelial cells. In another study, BJNhem19 and BJNhem20 stem cell lines were used to induce similar guided endodermal differentiation into pulmonary lineage cells. Both studies were done using similar protocols, where the hESCs were first plated on gamma irradiated mouse embryonic fibroblast (MEF) feeder cells and undifferentiated hESCs were taken through embroid body formation. They were then subjected to induction into lung specific cells, by defined growth factors in Small Airways Growth Medium (SAGM) and Bronchiolar Endothelial Growth Media (BEGM). The H7 cells were found to express, both intracellularly and on their surfaces, characteristic marker proteins. However, the BJNhem19 and BJNhem20 cells showed poor differential potential, and could not be successfully differentiated into lung- specific cells. This difference in differentiation potential may be due to several intrinsic limitations, like genetic heterogeneity, anomalous morphogen receptivity or dormant/ unresponsive cytoplasmic determinants. These observations indicate that the choice of cells may play an important factor in tissue engineering. This review addresses the different culture methods and conditions needed for guided differentiation of stem cells.
This perspective also discusses culture conditions of mesenchymal stem cells from different origins and dendritic stem cells from bone marrow.
Introduction
Stem cells are mother cells that have the potential to become any type of cell in the body. One of the main characteristics of stem cells is that they are undifferentiated cells capable of proliferation, self-renewal and differentiation towards specific phenotypes. Their differentiation is controlled by a variety of cues, including the nature of the substrate on which these cells lie, its innate stiffness, and the various mechanical forces at play in their surrounding microenvironments. These are known to be important in determining cell fate.
As one of the major components of regenerative medicine, tissue engineering follows the principles of cell transplantation, methods of bioengineering material science, toward the development of biologic substitutes that can restore, maintain normal function, and improve functions of a tissue or a whole organ following damage either by disease or trauma. Langer and Vacanti, 1993 .
The general principal of tissue engineering lies on 4 factors: scaffold, extracellular matrix (ECM), growth factors and cells Naughton, 2002 . Scaffold materials are three-dimensional tissue structures that guide the organization, growth and differentiation of cells. Growth factors are soluble peptides capable of binding cellular receptors and producing either a permissive or preventive cellular response toward differentiation and/or proliferation of tissue Naughton, 2002 . ECM must be capable of providing the optimal conditions for cell adhesion, growth, and differentiation within the construct by creating a system capable of controlling environmental factors such as pH, temperature, oxygen tension, and mechanical forces Whitaker et al., 2001 . These conditions are determined by the particular cell lines and the properties of the scaffold Whitaker et al., 2001 . Finally, the development of a viable construct involves a suitable supply of cells that are ideally non- immunogenic, highly proliferative, easy to harvest, and have the ability to differentiate into a variety of cell types with specialized functions Fuchs et al., 2001 Knight and Evans, 2004 Koh and Atala, 2004 Naughton, 2002 Shieh and Vacanti, 2005 Stock and Vacanti, 2001 .
The production of an engineered tissue in vitro requires the use of cells. Most of the successes in this field have come from the use of primary cells. However, this strategy has limitations, because of the invasive nature of cell collection and the potential for cells to be in a diseased state. Therefore, attention has become focused upon the use of stem cells, including embryonic stem (ES) cells, bone marrow mesenchymal stem cells (BM-MSCs) and umbilical cord-derived mesenchymal stem cells (UC-MSCs). Many stem cell lines are cultured on feeder cells to provide a conducive environment for growth, but there are implications for the transmission of xenogenic materials, so systems for growing stem cells in feeder-free systems are being established Denning et al., 2006 .
Our main aim is to ameliorate lung diseases, mainly idiopathic pulmonary fibrosis (IPF), by using stem cell tissue engineering based strategy. Most of the clinical therapies for pulmonary diseases are targeted towards improving the symptoms; there are no therapies that can cure intractable lung diseases. Therefore, we need a new approach to treat pulmonary diseases. Recent advances in stem cell research and tissue engineering, such as the establishment of inducible pluripotent stem (iPS) cells Takahashi and Yamanaka, 2006 open a new paradigm for future therapies of many intractable diseases. In contrast with these advances in other organs, stem cell research and tissue engineering in pulmonary diseases are limited. One reason is the complexity of the lung’s structure. The lung is a complex organ with a three-dimensional (3D) structure that is composed of over 40 cell types. Additionally, to mimic a functional lung, a perfect matching of gas and blood is needed. Therefore, the lung is the most challenging organ for tissue engineering Kubo, 2012 .
In this review we have focused on our own work and introduced a range of strategies and materials used for tissue engineering, including the sources of cells and cell lines suitable for this, like both mouse and human ESCs, BM-MSCs, adipose- derived MSCs (AD-MSCs), UC-MSCs and bone marrow derived dendritic cells (BMDCs).
Firstly, we will discuss about the culture and differentiation of human embryonic stem cells. Also called human pluripotent stem cells and derived from human embryos or fetal tissues, hESCs are self-replicating. They are known to develop into cells and tissues of the three primary germ layers (ectoderm, mesoderm, and endoderm), the primary layers of cells in the embryo from which all tissues and organs develop.Human embryonic stem cells have been used in a tissue engineering format to induce differentiation and amplification into the desired type of cell. As lung is our organ of interest, we have attempted to induce hESCs to differentiate into non- ciliated squamous epithelial cells.
Difference between cell lines
The promise of human embryonic stem cell (hESC) lines for treating injuries and degenerative diseases, for understanding early human development, for disease modelling and for drug discovery, has brought much excitement to scientific communities as well as to the public Allegrucci and Young, 2006 . Although all of the lines derived worldwide share the expression of characteristic pluripotency markers, many differences are emerging between lines that may be more associated with the wide range of culture conditions in current use than the inherent genetic variation of the embryos from which embryonic stem cells were derived, Allegrucci and Young, 2006 feeder layers that we used, growth media for maintaining embroid body, differentiation media and others. Thus, the validity of many comparisons between lines is difficult to interpret.
Here we deal with three cell lines- H7, BJNhem19, and BJNhem20. The h7 cell line was derived from the blastocyte of the frozen embryo by Thomson and his collaborators and it was differentiated into endodermal lineage, mainly lung lineage specific cells ( Figure 1 ) Ray Banerjee et. al 2012 . BJNhem19, and BJNhem20 were derived from the inner cell mass (ICM) of grade III poor quality blastocysts that were not suitable for in vitro fertility treatment, and we tried to differentiate them through guided endodermal differentiation into lung lineage specific cells ( Figure 2 ). However, the results were not satisfactory and the cells showed poor growth and practically none differentiated appreciably into the desired phenotype, although they were attempted to be grown under different conditions (with feeder, feeder-conditioned and feeder-free media). They were found not to grow in feeder free conditions, despite claim by the originator lab, and yield was not optimum even when grown on mouse feeder cells ( Figure 3 ). We even attempted to grow them with combinations of extra cellular matrix and defined medium (GeltrexR), as well as with enriched media such as Matrigel and mTeSR1 to no appreciable improvement. Of the cells that we obtained (sub-cultured up to passage 5) and experimented with, there was sequential down regulation of pluripotent markers- Oct ¾, SSEA-3 and SSEA-4 ( Figure 4 ), yet it did not yield satisfactory number of specific lineage differentiated cells nor maintain its pluripotent characters. From this standpoint we can say that heterogeneity is key to success of lineage specific differentiation of human embryonic stem cells.
Figure 1. Differentiation of H7 hESCs to lung-lineage specific cells
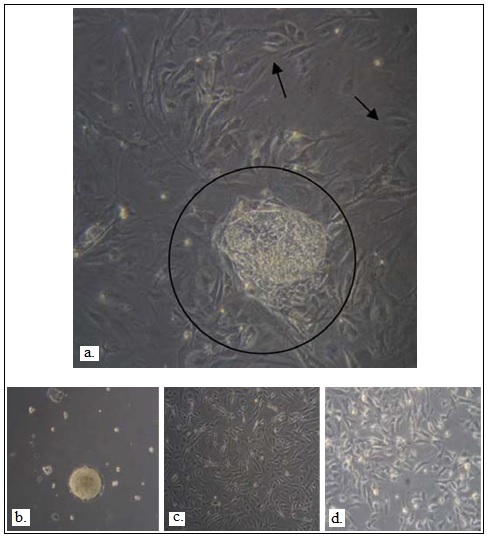
Figure 2. Differentiation of hESCs into lung- lineage specific cells
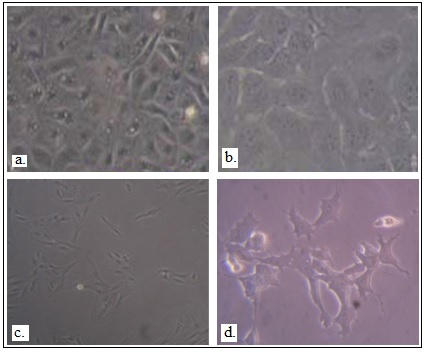
a. Phenotype of H7 in SAGM media. b. Phenotype of H7 in BEGM media. c. Phenotype of BJNhem 19 after culturing in SAGM medium. d. BJNhem 20 in BEGM media ( Garima Hore, 2015 Konsam Surajlata, 2015 ).
Figure 3. BJNhemembroid body formation in ultra- low attachment plates
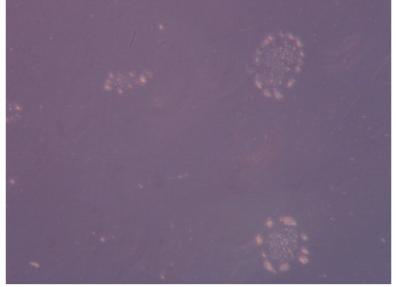
Figure 4. Expression of markers by BJNhem19, BJNhem20 and H7 cells in differentiation medium
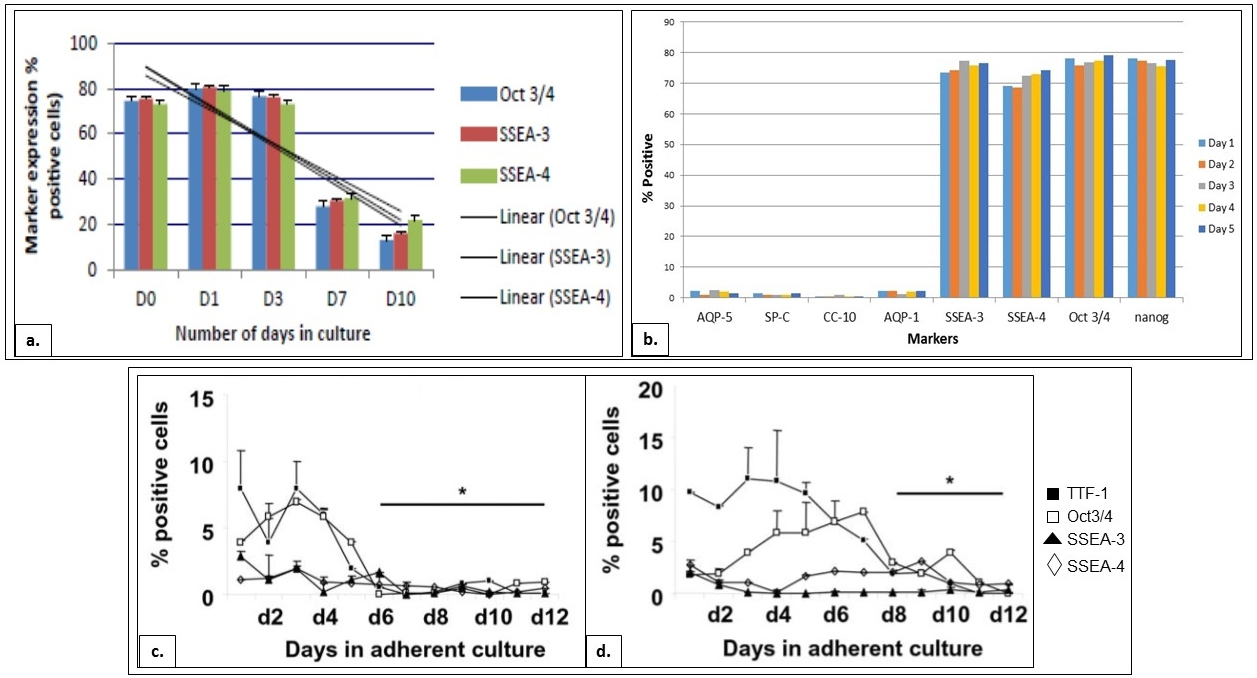
a. Sequential down- regulation of pluripotent markers on BJNhem19 over days in culture ( Garima Hore, 2015 Konsam Surajlata, 2015 ). b. Negligible down- regulation of pluripotent markers, and negligible expression of lung- specific markers, by BJNhem20 in differentiation medium. [Kar S., et al. 2014, 2015]. c. Down- regulation of pluripotent markers by H7 in SAGM ( Banerjee et al., 2012 )d. Down- regulation of pluripotent markers by H7 in BEG (Banerjee and Henderson, 2012).
Materials and methods
Cells- NIH approved undifferentiated H7 (human ESC line) was obtained from WiCell Research Institute, Wisconsin, USA. Human embryonic stem cell (hESC) lines BJNhem19 and BJNhem20 were obtained from JNCASR, Bangalore, India. Mesenchymal stem cells were isolated from mice bone marrow, adipose tissue and human umbilical cord, and dendritic cells from mice bone marrow.
Materials
Dulbecco’s modified eagle medium (DMEM), heat- inactivated fetal bovine serum (FBS), antibiotic- antimycotic (pen-strep) solution, 1X Trypsin- EDTA, and L- glutamine were obtained from Himedia, India. 1X DMEM and 1X Knockout TM DMEM F-12 (KO-DMEM) were obtained from Gibco, Life Technologies. SAGM was purchased from Promocell, Germany. Mitomycin C was obtained from Cayman Chemical Company. Triiodothyronine, Insulin, Bovine pituitary extract (BPE), retinoic acid, epinephrine, transferrin and hydrocortisone were bought from Lonza. Knockout serum replacement (KOSR), ESC- qualified FBS and dispase were obtained from Invitrogen, USA, and basic fibroblast growth factor (bFGF) from R & D Systems, USA. Sodium pyruvate and β- mercaptoethanol (β- ME) were bought from Sigma Aldrich Corporation, USA. Phosphate buffered saline (PBS), minimum essential medium (MEM) and non- essential amino acids (NEAA) were obtained from Mediatech, USA. Clonetics small airways basal medium and bronchiolar epithelial basal medium were purchased from Cambrex Bioscience, USA.
Antibodies were obtained from BD Pharmingen, USA, Santa Cruz Biotechnology, USA, or Chemicon, USA. 6 well ultralow attachment plates were obtained from Corning Inc. Lifesciences, USA.
Ethics Statement
All animal related work was conducted according to the guidelines prescribed by Faculty Councils for Post Graduate Studies in Science, Technology and Engineering and Agriculture & Veterinary Science (Ref. No. BEHR/1029/2304). Human embryonic stem cells (hESC) culture and differentiation protocol
Expansion of H7 hESCs
NIH approved (NIH code WA07) undifferentiated hES cell line H7 was obtained from WiCell Research Institute (Madison, WI), and cells from passage 25 to 35 were used. For propagation of the H7 cells in undifferentiated state, the ES cells were initially grown on primary mouse embryonic fibroblast (MEF) feeder cells prepared from timed pregnant CF-1 female mice (day 13.5 of gestation) that had been γ-irradiated with 3000 rads for 5 min, and then directly in conditioned medium in which the above γ-irradiated MEF cells were cultured to ensure purity of human cells and progressively eliminate any mouse feeders from the cultures. MEF cells were grown in DMEM, with 10% heat-inactivated FBS, and 2 mM L-glutamine. The hESCs were cultured in ESC medium (KO- DMEM supplemented with 20% KOSR, 1 mM sodium pyruvate, 0.1 mMβ-ME, 0.1 mM MEM, 1% NEAA, 1 mM L-glutamine, and 2 ng/ml bFGF. All cells were cultured on 6-well 10 cm 2 tissue culture plates, coated with 0.1% gelatin, and all cultures were incubated in a humidified incubator at 37°C with 5% CO 2 Ali et al., 2002 Banerjee and Henderson, 2012 Banerjee et al., 2012 Ray Banerjee and Henderson, 2009 Rippon et al., 2006 Samadikuchaksaraei et al., 2006 .
Expansion of Human embryonic stem cells (BJNhem19 and BJNhem20)
Human embryonic stem cell lines BJNhem19 and BJNhem20 were obtained from JNCASR, India. These cells were claimed to be karyotypically normal, sibling human ESC lines representing the Indian ethnic background. These cells were derived from the inner cell mass (ICM) of grade III poor quality blastocysts that were not suitable for in vitro fertility treatment. Both the lines as claimed by JNCASR were pluripotent, and have been extensively characterized and cultured continuously for over 250 passages Barbaric et al., 2011 Bilican et al., 2012 Bonig and Papayannopoulou, 2012 Garima Hore, 2015 Inamdar et al., 2011 Inamdar et al., 2009 Konsam Surajlata, 2015 Nestor et al., 2011 Rippon et al., 2006 Shetty and Inamdar, 2012 Venu et al., 2010 .
The aforementioned ESC lines were grown on primary mouse embryonic feeder cells, for expansion and propagation in an undifferentiated state. The feeder cells were prepared from pregnant Balb/C mice (13.5 days of gestation). These cells were then cultured in MEF conditioned media and treated with Mitomycin–c to stop their differentiation. The MEF was cultured in DMEM, with 10% FBS and 2 mM L- Glutamine Venu et al., 2010 . The hESCs were cultures in ES medium, comprising KO- DMEM supplemented with 20% KOSR, 1 mM sodium pyruvate, 0.1 mM 2β- ME, 0.1 mM MEM, 1% NEEA, 1 mM L-glutamine and 2 ng/ml bFGF. The cells were cultured on 6-well 10 cm 2 tissue culture plates, coated with 0.1% gelatin, and all cultures were incubated in a humidified incubator at 37°C with 5% CO 2 . The protocol for induction of alveolar epithelial differentiation of hESCs was adapted from established methods Banerjee, 2014 Banerjee and Henderson, 2013 .
Embroid Body (EB) Formation
On the day of passage, hESC colonies were inspected, and only hESC cultures containing colonies with well-defined boundaries and minimum differentiation were used. Undifferentiated hESCs were treated with 1.2 U/ml dispase dissolved in Ca 2+ +- and Mg 2+ +-free PBS, supplemented with 10% ESC-qualified FBS at 37°C until the hESC colonies nearly detached from the plates. Colonies were then washed off the plates, washed twice in ESC medium without bFGF, and resuspended in EB medium (i.e., KO DMEM, 20% KOSR, 20% non-heat-inactivated fetal calf serum, 1% NEAA, 1 mM L-glutamine, and 0.1 mMβ-ME). Cells were transferred to Corning 6-well ultra-low attachment plates, and grown for 4 days in suspension culture.
Generation of Non-ciliated Pulmonary Epithelial Cells
Two different culture media were employed to generate non-ciliated pulmonary epithelial cells. EBs were transferred to adherent culture in 0.1% gelatin-coated tissue culture plates by limited dispase digestion. One group of EBs was cultured for 12 days in small airways growth medium (SAGM) [Clonetics small airways basal medium, 30 µg/ml BPE, 5 µg/ml insulin, 0.5 µg/ml hydrocortisone, 0.5 µg/ml gentamycin sulfate-amphotericin B, 0.5 mg/ml bovine serum albumin, 10 µg/ml transferrin, 0.5 µg/ml epinephrine, and 0.5 ng/ml recombinant human epidermal growth factor (rh EGF)], refreshing media every other day. 0.1 ng/ml retinoic acid and 6.5 ng/ml triiodothyronine were excluded from SABM following Ali et al. From the day 12 culture in SAGM, alveolar epithelial cells were flow sorted based on surface expression of SP-C + and AQP-5 + . The >90% SP-C + + and AQP-5 + + flow-sorted cells were grown in the SAGM medium for 4 more days.
A second group consisted of EBs cultured in bronchiolar epithelial growth medium (BEGM) [Clonetics bronchiolar epithelial basal medium, 30 µg/ml BPE, 5 µg/ml insulin, 0.5 µg/ml hydrocortisone, 0.5 µg/ml gentamycin sulfate-amphotericin B, 0.1 ng/ml retinoic acid, 10 µg/ml transferrin, 6.5 ng/ml triiodothyronine, 0.5 µg/ml epinephrine, and 0.5 ng/ml rh EGF], and fed similarly every other day with fresh medium.
Phenotypic Analysis of Cells
Immunostaining was performed using specific antibodies conjugated to various fluorochromes such as fluorescein isothiocyanate (FITC), phycoerythrin (PE), allophycocyanin (APC), peridinin chlorophyll protein (PerCP-Cy5.5), and CyChrome (PE-Cy5 and PE-Cy7). The following BD Biosciences Pharmingen (San Diego, CA) antibodies were used for cell surface staining: APC-conjugated CD45 (30F-11), FITC-conjugated CD3 (145-2C11), PE-Cy5-conjugated B220 (RA3-6B2), APC-conjugated GR-1 (RB6-8C5), PE-conjugated Mac1 (M1/70), FITC-conjugated Sca-1, and PE-Cy7-conjugated CD117 (c-kit). PE-Cy5-conjugated F4/80 [Cl: A3-1 (F4/80)] was obtained from Serotec Ltd., Oxford, UK. Purified antibodies (clone number/catalog number/antibody type/concentration) to the following mouse antigens were obtained from Santa Cruz Biotechnology (Santa Cruz, CA): SSEA-3 (631/sc-21703/rat monoclonal IgM/200 µg/ml), SSEA-4 (813-70/sc-21704/mouse monoclonal IgG 3 /200 µg/ml), Oct-3/4 (H-134/sc-7705/goat polyclonal IgG/200 µg/ml), SP-C + (C-19/sc-7705/goat polyclonal IgG/200 µg/ml), SP-D (245-01/sc-59695/mouse monoclonal IgG1/100 µg/ml), AQP-1 (L-19/sc-9878/goat polyclonal IgG/200 µg/ml), AQP-5 + (G-19/sc-9890/goat polyclonal IgG/200 µg/ml), CC-10 (S-20/sc-9773/goat polyclonal IgG/200 µg/ml), EGF (C-20/sc-1341/goat polyclonal IgG/200 µg/ml, VEGF (P-20/sc-1836/goat polyclonal IgG/200 µg/ml), TTF-1 (G-17/sc-12524/goat polyclonal IgG/200 µg/ml), CD31 [i.e., platelet endothelial cell adhesion molecule (PECAM-1); V-16/sc-31045/goat polyclonal IgG/200 µg/ml], and goat anti-mouse IgG 3 -FITC (sc-2081/pre-adsorbed, affinity-purified secondary antibody raised in goat against mouse IgG 3 and conjugated to FITC/400 µg/ml). Irrelevant isotype-matched antibodies were used as controls. FITC-conjugated donkey anti-goat or goat anti-rabbit secondary antibodies were used following incubation with the primary antibodies. In situ immunostaining with specific FITC- or PE-conjugated antibodies (and DAPI counterstaining the nuclei of the cells) or ABC staining (DAKO) was done following the manufacturer's protocol. 10 6 cells were taken per sample in 50 µl cell suspension in ice cold 1X PBS; 10 5 events were recorded per sort.
Fluorescein-activated cell sorter (FACS) analysis
For simultaneous surface and intracellular staining, cell-surface antigens were stained as follows: 1 µl conjugated antibody/10 6 cells in suspension culture for 30 min on ice. After thorough washing, cells were fixed in 4% paraformaldehyde in PBS by vortexing, and incubated at room temperature (RT) for 20 min followed by permeabilization in either 0.1% Tween-20 or 0.25% Triton-X. Intracellular staining was performed with readouts made on a BD FACScalibur. Different conjugates with widely separated excitation spectral range were used for separating the surface vs. intracellular probes (e.g., PE vs. FITC, FITC vs. APC, APC vs. PE, or APC vs. Cyc-PE).
Cell suspension of 10 6 cells per microfuge tube was prepared per sample and staining was done by a single step with a master mix of fluorochrome-conjugated monoclonal antibodies or in some cases where the primary antibody was not available in a directly fluorochrome-conjugated form, in two steps of primary unlabeled antibody followed by cross reactive fluorochrome-conjugated specific secondary antibody at 4°C for 30 min followed by rigorous washing (twice) with ice cold PBS. The stained cell preparation was finally resuspended in 50 µl PBS (with 1% bovine serum albumin) and read by FACSCalibur (BD Immunocytometry Systems, San Jose, CA) by using the CELLQuest program. Cells were viewed at first keeping at Side Scatter (SSC; X-axis) and Forward Scatter (FSC; Y-axis) and dead cells gated out by annexin V staining. CD45− cells were then gated out to preempt any blood cells in the lungs, and 10 5 events were recorded per sample. The unstained axis was FL-3H. In undifferentiated H7 cells, single staining with each antibody was done for TTF-1, Oct3/4, SSEA-3, and SSEA-4. Data from three independent experiments with each sample sorted in triplicate were pooled, and mean ± SEM reported.
Cell Viability
Viable cells were measured by propidium iodide exclusion using flow cytometry and trypan blue dye exclusion by light microscopy.
Clonogenic Growth of Cells Derived from hES Cells
To quantitate committed progenitors, CFU-c assays were performed using methylcellulose semisolid media (Stemgenix, Amherst, NY) supplemented with an additional 50 ng of stem cell factor per ml (Peprotech, Rocky Hill, NJ) to promote growth of hematopoietic progenitors. 0.01×10 6 cells from lung were plated in duplicate in 35-mm culture dishes and incubated at 37°C in a 5% CO 2 -95% air mixture in a humidified chamber for 7 days. Colonies generated by that time were counted using a dissecting microscope, and all colony types (i.e., BFU-E, CFU-E, CFU-G, CFU-GEMM, CFU-GM, and CFU-M) were pooled and reported as total CFU-c. Aliquots of 1–10×10 4 cells were plated per 1 ml of semisolid methylcellulose (CFU-lite with Epo, Miltenyl Biotech, or complete human methycellulose medium, Stem Cell Technologies, Vancouver, BC, Canada). CFU-c frequency was scored morphologically after 10 to 14 days in culture at 37°C, and 5% CO 2 , in a humidified incubator.
RNA Isolation
Total RNA was extracted from cultured cells (<500/sample) by PicoPure RNA isolation kit (Arcturus, Mountain View, CA). For isolation of total lung RNA, 600 µl of lysing buffer was added to disrupted lung tissue in a 1.5-ml microfuge tube, and lysate was loaded onto a QIA shredder column and centrifuged for 2 min at 13,000 rpm. The homogenized lysate was then mixed with 600 µl of 70% ethanol and applied to an RNeasy mini spin (QIAGEN Inc, Valencia, CA) column for centrifugation for 15 sec at 13,000 rpm. Next, 700 µl of buffer RW1 and buffer RPE was added and centrifuged sequentially for washing twice. Then, 60 µl of ribonuclease-free water was used to elute total RNA from the RNeasy mini spin column. All total RNA used in the experiments was pure as determined by the ratio of absorbance (A) at 260 vs. 280 nm (A260/A280 ratio >1.9) and stored at −80°C.
Quantitative PCR (qPCR) Analysis
cDNA was made using Superscript III system from Invitrogen and qPCR performed. For qPCR performed in duplicate tubes, the PCR reaction solution contained 0.5 µg of total RNA, 6 mM magnesium chloride, and 0.5 µM of each primer. Other components in the reverse transcriptase PCR master mix included buffer, enzyme, SYBR Green I, and deoxyribonucleotide triphosphate. For reverse transcription, the 20 µl of reaction capillaries were incubated at 50°C for 2 min followed by denaturation at 95°C for 10 min. PCR by initial denaturation at 95°C for 15 sec was followed by annealing at 60°C for 1 min, repeated 45 cycles. Finally, a melting curve analysis was performed by following the final cycle with incubation at 95°C for 15 sec, 60°C for 15 sec, and 95°C for 15 sec. Negative control samples for the qPCR analysis that contained all reaction components except RNA, were performed simultaneously to determine when the nonspecific exponential amplification cycle number was reached. Primers were synthesized by the University of Washington Biochemistry services using Primer Express software. qPCR was performed by the comparative Ct method with SYBR Green PCR core reagents (Applied Biosystems, Foster City, CA) and analyzed using Applied Biosystems 7900HT Real-Time PCR System software SDS 2.2.1.
Validation of tissue engineered differentiated cells in models of differentiation
Bleomycin model is our injury model for developing fibrosis. During bleomycin induced lung injury, endothelial cells and AEI cells are the initial sites of damage. This is followed by interstitial edema and increased deposition of extracellular matrix proteins, such as collagen, lamin and fibronectin, fibroblast recruitment, proliferation, and differentiation. This results in scarring of the lung, architectural distortion, and irreversible loss of function. To ameliorate this condition, we used cell based regenerative therapy and tried to differentiate hESCs to pulmonary lineage specific cells, to repair lung injury. For this, we used the following methods.
Analysis of Collagen Content in Lung
Masson's trichrome and Sirius red stains were used to detect collagen deposition in the lungs. Total amount of soluble collagen in the lung was determined as the mean of triplicate tubes for each sample by the Sircol™ quantitative dye-binding collagen assay (Biocolor Ltd., Newtownabbey, Northern Ireland, UK) Teo et al., 2005 .
Detection of Human Cells in Mouse Lung
Three methods were employed to detect engrafted cells in mouse lung:
1) Detection of Alu sequence in transplanted mouse lung RNA was performed by qPCR, using the following primers: GTCAGGAGATCGAGACCATCCC (forward sequence) and TCCTGCCTCAGCCTCCCAAG (reverse sequence). Alu elements are specific to the human genome and are present at ~1 million copies/diploid sequence, making them a sensitive indicator of human cell content.
2) Immunohistochemistry was performed using a mouse anti-human nuclei IgG1 monoclonal antibody (clone 235-1, catalog number: MAB1281; Millipore Corporation, Billerica, MA), that stains nuclei of all human cell types giving a diffuse nuclear pattern with no reactivity against mouse in immunohistochemistry. 5 µm thick sections of 2% paraformaldehyde-fixed OCT-embedded frozen lung tissues were blocked with goat serum at RT for 1 h, followed by incubation with the anti-human nuclei antibody overnight at 4°C. After washing in PBS and incubation with goat anti-mouse secondary antibody for 1 h at RT, ABC staining (Vector Laboratories Inc.) was performed following the manufacturer's protocol.
3) In situ hybridization in mouse transplanted lung sections with human-specific pan-centromeric probe was performed in 8 µm thick, methyl carnoy-fixed paraffin-embedded lung sections following the protocol described previously Laflamme et al., 2005 .
Immunohistochemistry
For immunohistochemistry with non-conjugated antibodies, paraffin-embedded lung tissue was deparaffined in xylene, and rehydrated in 100% and 95% ethyl alcohol. Endogenous peroxidase was quenched in methanol with 0.3%–3% hydrogen peroxide for 30 min at RT. Blocking was done for 1 h at RT in PBS containing Ca 2+ + and Mg 2+ + with 1.5% non-immune serum of the species in which the secondary antibody was made. The primary antibody was incubated for 1 h at RT followed by 3 washes in PBS at RT. The secondary antibody was applied and incubated. ABC staining (Vector Laboratories Inc, Burlingame, CA) was performed following the manufacturer's protocol.
Immunofluorescence Microscopy
Photographs were taken with a Leica DMIL inverted microscope (Leica Microsystems GmbH, Wetzlar, Germany) and a Zeiss ApoTome (Carl Zeiss Microimaging GmbH, Göttingen, Germany). IF photographs were taken with a Zeiss Axiovert 200 M microscope and AxiocamMRm and merged using Axiovision 4.6 software.
Transmission electron microscopy
For transmission electron microscopy, the cells were fixed with warm ½Karnovsky's fixative (1:1 with buffer) after removal of the culture medium and washed with 0.1 M cacodylate buffer for 10 min. After the fixative was removed, the sections were incubated in pure fixative for 30–60 min. The cells were gently scraped using a standard Sarstedt cell scraper, and placed into Eppendorf tubes and spun down at 1500 rpm for 5 min. After addition of new fixative, the cells were resuspended and stored at 4°C overnight. After 3 washes for 5 min in 0.1 M cacodylate buffer, cells were centrifuged and 1% osmium tetroxide in 0.1 M cacodylate buffer added and incubated for 1–2 h at 4°C. This was followed by 3 washes in 0.1 M cacodylate buffer for 5 min. Dehydration was done in graded series of ethyl alcohol (i.e., 50%, 70%, 95%, 2×100%) for 15 min and two washes in propylene oxide for 15 min. Embedding was done in 1:1 propylene oxide/Epon resin overnight with Eppendorf tubes capped. The next day, cells were centrifuged and fresh 100% Epon resin added for 2–4 h. Polymerization was done in a 60°C oven overnight in Eppendorf tubes. 70–100 nm thick sections were made on a copper grid using a Leica EM UC6 ultramicrotome (Leica Microsystems GmbH, Wetzlar, Germany). Sections were viewed in a JEOL JEM-1230 transmission electron microscope (JEOL Ltd., Tokyo, Japan), equipped with an Ultrascan 1000™ 2k×2k CCD camera (Gatan, Inc., Pleasanton, CA), and photomicrographs taken using Gatan Digital Microscope software.
Mesenchymal stem cell isolation and culture protocol
Mesenchymal stem cells (MSCs) are multipotent stromal cells that have a property of differentiation into a variety of cell types. Those cells include osteoblast or bone cells, chondrocytes or cartilage cells, and adipocytes or fat cells. MSCs do not differentiate into hematopoietic cells. They can be derived from bone marrow or other non- marrow tissues, such as adipose tissue, adult muscle, corneal stroma, or dental pulp of baby, because they have the capability to regenerate different tissue However, as they do not have the capacity to reconstitute an entire organ, the term ‘multipotent stromal cells’ has been proposed as a better replacement.
The most primitive form of MSC can be isolated from the umbilical cord tissue, namely Wharton jelly, and the umbilical cord blood. The MSCs are found in much higher concentration in the Wharton jelly than in the umbilical cord blood, which is, on the other hand, a source of hematopoietic stem cells. The UC- MSCs have more primitive properties than adult MSCs obtained later in life, which makes them a good source of MSC for clinical application Ray Banerjee, 2014 .
Figure 5. Transmission electron microscopy of cells differentiated from H7 stem cells
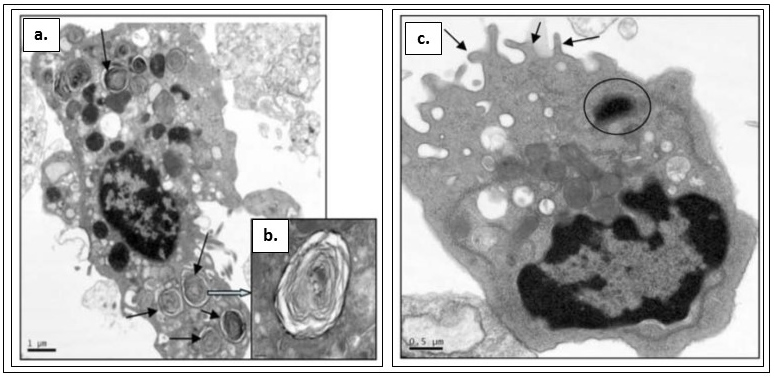
a. AEII cells grown in SAGM. b. AEII cells grown in SAGM (close- up image of a.). c. Clara cell grown in BEGM ( Banerjee et al., 2012 ).
Figure 5. Transmission electron microscopy of cells differentiated from H7 stem cells
a. AEII cells grown in SAGM. b. AEII cells grown in SAGM (close- up image of a.). c. Clara cell grown in BEGM ( Banerjee et al., 2012 ).
Isolation of human umbilical cord MSC (hUC- MSC)
Human umbilical cord was collected from hospital with the consent of parents. Umbilical cord was stored in DMSO at – 80 0C in a 50 ml centrifuge tube. On the day of experiment, the tube was thawed at 37 0C quickly till few ice crystals remain. The cord was quickly washed in DMSO neutralizing media (DMEM + 10% FBS) and transferred into a Petri dish containing 7 ml growth medium (DMEM + 10% FBS + 1% Pen-Strep). The inner content of the UC was squeezed out into the media using forceps and scalpel. The remaining cord was chopped into small sections using surgical blades, and the sections were opened up for better interaction with growth medium. Sections were placed in a 100-cm tissue culture plate and 20- ml media was added. The inner content containing media (step -4) was added to the plate and the entire plate was kept at 370 C incubation (with 5% CO 2 ) for 5 days. The setup was kept undisturbed for 3 days and then observed under microscope for any adherent cells. Setup was replaced in incubator for further growth. Once cell becomes confluent they were passaged; cells were detached using trypsin EDTA. For storage of cells, cell suspension was transferred into a cryovial containing freezing medium (10% DMSO and 90% media with FBS) and stored.
Isolation and culture of mesenchymal stem cells from murine bone marrow (mBM- MSCs)
Mouse MSCs are generally isolated from an aspirate of bone marrow harvested from the tibia and femoral marrow compartment, then cultured in DMEM with FBS, for 3 hrs in 370C, at 5% CO 2 . Non adherent cells were removed carefully after 3 hrs and fresh medium was added. When primary cultures became almost confluent, the culture was treated with 0.5 ml of 0.25% trypsin EDTA for 2 min at room temperature (250C). A purified population of MSCs can be obtained after the initiation of culture Soleimani and Nadri, 2009 .
Isolation and culture of mesenchymal stem cells from murine adipose tissue (mAD- MSCs)
Inguinal adipose tissues of 12–14 weeks old Balb/c mice were isolated and digested at 37°C in PBS containing 2% bovine serum albumin (BSA) and 2 mg/ml collagenase (collagenase A, Roche), for 15–20 min. After filtration through 40 μm nylon filter mesh (BD Falcon) and centrifugation, isolated cells were suspended in medium, counted with a hemocytometer, plated at 5 × 10 4 cells/ml in 6 cm tissue culture plates, and cultured DMEM with 20% FBS. The cells were daily observed under an inverted phase-contrast microscope and were passaged after 80–90% confluence. The culture media was changed every two days Taha and Hedayati, 2010 .
Dendritic cell isolation and culture protocol from murine bone marrow (mBMDCs)
Dendritic cells (DCs) are antigen-presenting cells (APCs) which play a critical role in the regulation of the adaptive immune response. Also referred to as professionals APCs, they have the ability to induce a primary immune response in resting naïve T lymphocytes. DCs are capable of capturing antigens, processing them, and presenting them on the cell surface along with appropriate costimulation molecules. Dendritic cells can be isolated from mouse spleen and from other mouse tissue as well as from human tissues. Here we have discussed about the isolation and culture of dendritic cells from murine bone marrow.
Isolation of dendritic cells
8-10 week male C57BL6 mice were euthanized by CO 2 asphyxiation. The hind legs were cut, cleaned with ethanol and collected in RPMI- 1640 medium. The ends were trimmed, and media was flushed through the bone to collect the bone marrow in centrifuge tubes. The cell suspension was diluted, centrifuged at 250g for 8 minutes. The pellet was washed twice with HBSS, and resuspended in culture medium (RPMI-1640, 10% FBS, 20 mM pen/ strep) to a final cell density of 10 x 10 6 . The cells were then plated in 90 mm Petri dishes, to a density of 2 x 10 6 per plate. 0.2 ml of 1000 ng/ml recombinant murine granulocyte macrophage colony stimulating factor (rmGM-CSF) was added into above Petri dishes, so that final concentration of rmGM-CSF in 10 ml is 20 ng/ml. The contents were mixed uniformly, and incubated at 37°C, 5% CO 2 and 95% humidity for 3 days.
Primary BMDCs were harvested form each Petri dish by collecting the non- adherent cells. The loosely adherent and non- adherent cells were collected in centrifuge tubes, and the adherent cells were discarded, as they contained macrophages. The cells were centrifuged, and resuspended in RPMI-1640 medium, at a density as required for assays Madaan et al., 2014 .
Discussion
Our observations have shown that all ESCs are not successfully differentiated into a particular lineage. Where H7 ESCs could be differentiated into lung- lineage specific cells, BJNhem19 and BJNhem20 ESCs could not be differentiated by the same method. It has been observed by other groups of investigators that differentiation of stem cells can be guided by the choice of medium or extracellular matrix Li et al., 2013 Santiago et al., 2009 . It was found that interaction of MSCs with the ECM protein can guide the cells towards multiple lineages, like fat, bone, or muscle Santiago et al., 2009 . Another group has successfully differentiated UC-MSCs into odontoblast-like cells when grown in conditioned medium for tooth germ cells Li et al., 2013 .
Artificial transplantation is a successful therapy for otherwise incurable end-stage diseases or tissue loss. However, such interventions are challenged by organ shortage, the necessity of lifelong immunosuppression and its potential for serious complications. Tissue engineering has emerged as a rapidly expanding approach to address these problems and is a major component of regenerative medicine Fuchs et al., 2001 Knight and Evans, 2004 Shieh and Vacanti, 2005 .
The field of regenerative medicine and tissue engineering is an ever evolving field that holds promise in treating numerous diseases and injuries. An important aspect in the development of the field was the discovery and implementation of stem cells. The utilization of mesenchymal stem cells, and later embryonic and induced pluripotent stem cells, opens new arenas for tissue engineering and presents the potential of developing stem cell-based therapies for disease treatment. The overarching goal of stem cell-based tissue engineering research is to precisely control differentiation of stem cells in culture and maintain their heterogeneity T. Brown et al., 2013 .
Recent advances in the field of regenerative medicine provide us with a unique set of tools to improve the quality of engineered tissue. The use of engineered embryonic stem cells differentiated into tissue specific cells and specific adult stem cells, assembly through additive manufacturing and improved understanding of postnatal tissue maturation will allow us to more accurately replicate native tissue anisotropy. There are however many limitations to autologous tissue engineering and transplantation in regenerative medicine including donor site morbidity, technical considerations and long term complications that must be addressed. Current tissue engineered organ constructs implanted into immune-competent animal models have been observed to undergo inflammation, fibrosis, foreign body reaction, calcification and degradation. Combining biomimetic regenerative medicine strategies will allow us to improve tissue engineered lung lineage specific cells with respect to biochemical composition and functionality, as well as microstructural organization and overall shape, for successful trouble free long term integration into viable functional tissue. Creating functional and durable tissue has the potential to shift the paradigm in reconstructive surgery by obviating the need for donor sites.
Although tissue engineering is in its relative infancy, huge advances are being made through the collaborations between stem cell biologists and material chemists. Identification of novel stem cell populations, with unique properties will improve this process. It is hoped that by combining stem cell technologies with materials able to deliver combinations of growth factors, we may be able to treat conditions requiring reconstructive surgery or organ replacement. The growth of knowledge in both stem cell biology, through the investigation of the differentiation process, and materials science, by development of novel polymers and scaffold structures, is bringing the time of routine engineered tissues closer Howard et al., 2008 .
References
- N.N. Ali, A.J. Edgar, A. Samadikuchaksaraei, C.M. Timson, H.M. Romanska, J.M. Polak, A.E. Bishop. Derivation of Type II Alveolar Epithelial Cells from Murine Embryonic Stem Cells. Tissue Engineering. 2002;8:541-550. Google Scholar
- C. Allegrucci, L.E. Young. Differences between human embryonic stem cell lines. Human Reproduction Update. 2006;13:103-120. Google Scholar
- E. Banerjee. Looking for the elusive lung stem cell niche. Transl Respir Med. 2014;2:7. Google Scholar
- E. Banerjee, W. Henderson. Characterization of lung stem cell niches in a mouse model of bleomycin-induced fibrosis. Stem Cell Research & Therapy. 2012;3:21. Google Scholar
- E.R. Banerjee, W.R. Henderson. Role of T cells in a gp91phox knockout murine model of acute allergic asthma. Allergy, Asthma & Clinical Immunology. 2013;9:6. Google Scholar
- E.R. Banerjee, M.A. Laflamme, T. Papayannopoulou, M. Kahn, C.E. Murry, W.R. Henderson. Human Embryonic Stem Cells Differentiated to Lung Lineage-Specific Cells Ameliorate Pulmonary Fibrosis in a Xenograft Transplant Mouse Model. PLoS ONE. 2012;7:e33165. Google Scholar
- I. Barbaric, M. Jones, K. Buchner, D. Baker, P.W. Andrews, H.D. Moore. Pinacidil enhances survival of cryopreserved human embryonic stem cells. Cryobiology. 2011;63:298-305. Google Scholar
- B. Bilican, A. Serio, S.J. Barmada, A.L. Nishimura, G.J. Sullivan, M. Carrasco, H.P. Phatnani, C.A. Puddifoot, D. Story, J. Fletcher. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proceedings of the National Academy of Sciences. 2012;109:5803-5808. Google Scholar
- H. Bonig, T. Papayannopoulou. Mobilization of Hematopoietic Stem/Progenitor Cells: General Principles and Molecular Mechanisms. In Stem Cell Mobilization (Springer Science + Business Media). 2012;:1-14. Google Scholar
- C. Denning, C. Allegrucci, H. Priddle, M.D. Barbadillo-munoz, D. Anderson, T. Self, N.M. Smith, T. Parkin, L.E. Young. Common culture conditions for maintenance and cardiomyocyte differentiation of the human embryonic stem cell lines, BG01 and HUES-7. Int J Dev Biol. 2006;50:27-37. Google Scholar
- J.R. Fuchs, B.A. Nasseri, J.P. Vacanti. Tissue engineering: a 21st century solution to surgical reconstruction. The Annals of Thoracic Surgery. 2001;72:577-591. Google Scholar
- S.K. Garima Hore. Human Embryonic Stem Cell Lines BJNhem 19 and 20 Fail to Differentiate Into Lung Lineage Specific Cells despite Induction through Guided Endodermal Differentiation. J Tissue Sci Eng. 2015;06:. Google Scholar
- D. Howard, L.D. Buttery, K.M. Shakesheff, S.J. Roberts. Tissue engineering: strategies, stem cells and scaffolds. Journal of Anatomy. 2008;213:66-72. Google Scholar
- M.S. Inamdar, L. Healy, A. Sinha, G. Stacey. Global Solutions to the Challenges of Setting up and Managing a Stem Cell Laboratory. Stem Cell Rev and Rep. 2011;8:830-843. Google Scholar
- M.S. Inamdar, P. Venu, M.S. Srinivas, K. Rao, K. VijayRaghavan. Derivation and Characterization of Two Sibling Human Embryonic Stem Cell Lines From Discarded Grade III Embryos. Stem Cells and Development. 2009;18:423-434. Google Scholar
- M.A.F. Knight, G.R.D. Evans. Tissue Engineering: Progress and Challenges. Plastic and Reconstructive Surgery. 2004;114:26e-37e. Google Scholar
- C.J. Koh, A. Atala. Therapeutic Cloning and Tissue Engineering. In Current Topics in Developmental Biology (Elsevier BV),. 2004;:1-15. Google Scholar
- S.K. Konsam Surajlata. Heterogeneity in Human Embryonic Stem Cells May Prevent Endodermal Guided Differentiation. Journal of Stem Cell Research & Therapy. 2015;05:. Google Scholar
- H. Kubo. Concise Review: Clinical Prospects for Treating Chronic Obstructive Pulmonary Disease with Regenerative Approaches. Stem Cells Translational Medicine. 2012;1:627-631. Google Scholar
- M.A. Laflamme, J. Gold, C. Xu, M. Hassanipour, E. Rosler, S. Police, V. Muskheli, C.E. Murry. Formation of Human Myocardium in the Rat Heart from Human Embryonic Stem Cells. The American Journal of Pathology. 2005;167:663-671. Google Scholar
- R. Langer, J. Vacanti. Tissue engineering. Science. 1993;260:920-926. Google Scholar
- T.X. Li, J. Yuan, Y. Chen, L.J. Pan, C. Song, L.J. Bi, X.H. Jiao. Differentiation of Mesenchymal Stem Cells from Human Umbilical Cord Tissue into Odontoblast-Like Cells Using the Conditioned Medium of Tooth Germ CellsIn Vitro. BioMed Research International. 2013;2013:1-10. Google Scholar
- A. Madaan, R. Verma, A.T. Singh, S.K. Jain, M. Jaggi. A stepwise procedure for isolation of murine bone marrow and generation of dendritic cells. J Biol Methods. 2014;1:. Google Scholar
- G.K. Naughton. From Lab Bench to Market. Annals of the New York Academy of Sciences. 2002;961:372-385. Google Scholar
- C.E. Nestor, R. Ottaviano, J. Reddington, D. Sproul, D. Reinhardt, D. Dunican, E. Katz, J.M. Dixon, D.J. Harrison, R.R. Meehan. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Research. 2011;22:467-477. Google Scholar
- E. Ray Banerjee. Perspectives in Regenerative Medicine. Springer Science + Business Media. 2014;:. Google Scholar
- E. Ray Banerjee, J.W.R. Henderson. Characterization of Lung Stem Cell Niches in a Mouse Model of Bleomycin-Induced Fibrosis. In A58 LUNG STEM AND PROGENITOR CELLS (American Thoracic Society). 2009;:. Google Scholar
- H.J. Rippon, J.M. Polak, M. Qin, A.E. Bishop. Derivation of Distal Lung Epithelial Progenitors from Murine Embryonic Stem Cells Using a Novel Three-Step Differentiation Protocol. Stem Cells. 2006;24:1389-1398. Google Scholar
- A. Samadikuchaksaraei, S. Cohen, K. Isaac, H.J. Rippon, J.M. Polak, R.C. Bielby, A.E. Bishop. Derivation of Distal Airway Epithelium from Human Embryonic Stem Cells. Tissue Engineering. 2006;12:867-875. Google Scholar
- J.A. Santiago, R. Pogemiller, B.M. Ogle. Heterogeneous Differentiation of Human Mesenchymal Stem Cells in Response to Extended Culture in Extracellular Matrices. Tissue Engineering Part A. 2009;15:3911-3922. Google Scholar
- R. Shetty, M.S. Inamdar. Derivation of Human Embryonic Stem Cell Lines from Poor Quality Embryos. In Methods in Molecular Biology (Springer Science + Business Media). 2012;:151-161. Google Scholar
- S.-J. Shieh, J.P. Vacanti. State-of-the-art tissue engineering: From tissue engineering to organ building. Surgery. 2005;137:1-7. Google Scholar
- M. Soleimani, S. Nadri. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat Protoc. 2009;4:102-106. Google Scholar
- U.A. Stock, J.P. Vacanti. Tissue Engineering: Current State and Prospects. Annual Review of Medicine. 2001;52:443-451. Google Scholar
- P. T. Brown, A. M. Handorf, W. Bae Jeon, W.-J. Li. Stem Cell-based Tissue Engineering Approaches for Musculoskeletal Regeneration. CPD. 2013;19:3429-3445. Google Scholar
- M.F. Taha, V. Hedayati. Isolation, identification and multipotential differentiation of mouse adipose tissue-derived stem cells. Tissue and Cell. 2010;42:211-216. Google Scholar
- K. Takahashi, S. Yamanaka. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663-676. Google Scholar
- J.L. Teo, H. Ma, C. Nguyen, C. Lam, M. Kahn. Specific inhibition of CBP/ -catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proceedings of the National Academy of Sciences. 2005;102:12171-12176. Google Scholar
- P. Venu, S. Chakraborty, M.S. Inamdar. Analysis of long-term culture properties and pluripotent character of two sibling human embryonic stem cell lines derived from discarded embryos. In Vitro Cellular & Developmental Biology - Animal. 2010;46:200-205. Google Scholar
- M.J. Whitaker, R.A. Quirk, S.M. Howdle, K.M. Shakesheff. Growth factor release from tissue engineering scaffolds. Journal of Pharmacy and Pharmacology. 2001;53:1427-1437. Google Scholar



![[Download figure]](psc.v3i03.127/fig1.png){kind=link}
![[Download figure]](psc.v3i03.127/fig2.png){kind=link}
![[Download figure]](psc.v3i03.127/fig3.png){kind=link}
![[Download figure]](psc.v3i03.127/fig4.png){kind=link}
![[Download figure]](psc.v3i03.127/fig5.png){kind=link}
a. Undifferentiated hES cells (within circle) were expanded on c-irradiated MEF feeders for 4–6 days followed by formation of b. b. EBs in suspension culture overnight after aggregation. c. Day 4 EBs were cultured in ultra-low attachment plates for 10 days and then transferred to gelatin-coated plates, and cultured in SAGM d. Day 4 EBs were cultured in ultra-low attachment plates for 10 days and then transferred to gelatin-coated plates, and cultured in BEGM ( Banerjee et al., 2012 ).